单细胞RNA测序(scRNA-seq)技术已经成为揭示单个细胞内RNA转录本的异质性和复杂性,以及揭示组织/器官/生物体内不同细胞类型的组成和功能的最先进的方法。自2009年首次发现以来,基于scRNA-seq的研究提供了不同领域的大量信息,在更好地理解人类、模式动物和植物内细胞的组成和相互作用方面取得了众多令人兴奋的新发现。
近日青岛-欧洲生命科学高等研究院科研团队在Clinical and Translational Medicine杂志(影响因子:11.492)发表最新综述,简要概述了scRNA-seq技术、实验和计算方法将生物学和分子过程转化为计算和统计数据。并对实现该技术的关键技术步骤进行了说明。作者着重举例说明了scRNA-seq如何为更好地理解健康和疾病提供独特的信息。scRNA-seq技术的一个重要应用是建立更好和高分辨率的所有生物体的细胞目录,通常被称为图谱,可以作为理解和提供治疗疾病的解决方案的关键资源。虽然该技术在所有领域都显示了巨大的前景,但作者也强调了一些尚待克服的挑战,以及它在转变当前疾病诊断和治疗方案方面的巨大潜力。

人体是高度有序的系统,由大约3.72×1013个不同类型的细胞组成,形成和谐的微环境以保持正常的器官功能和正常的细胞动态平衡。活细胞在16世纪第一次被发现,从那时起,许多新技术和方法从简答到复杂逐步发展起来。尽管16世纪晚期Zacharias Janssen和Hans Lippershey发明的第一台显微镜使Robert Hooke和Anton van Leeuwenhoek在17世纪发现了第一个活细胞,花费了将近两个世纪来重新定义细胞不仅是生命的结构单位,而且是生命的功能单位。
此后,开展了各种实验和方法为了更好地理解和研究异质性多细胞系统中的细胞。尽管在细胞生物学领域已经有了巨大的革命性的发现,但细胞的异质性仍有待进一步研究。人体中几乎所有的细胞都有一组相同的遗传物质,但每个细胞中的转录组信息反映了一部分基因的独特活性。分析细胞内的基因表达活性被认为是探测细胞身份、状态、功能和反应的最可靠的方法之一。在过去的十年中,单细胞转录组学取得了巨大的技术突破。通过单细胞RNA测序,现在可以在单个研究中在单细胞水平分析数百万个细胞的转录组。这使我们能够在转录组水平上对每个细胞进行分类、表征和区分,从而识别出罕见但功能重要的细胞群。
汤富酬等人于2009年首次在概念上和技术上取得单细胞RNA测序方法的突破,对单个卵裂球和卵母细胞的转录组进行了测序。这项研究带来的概念和技术为扩大细胞数量开辟了一条新途径,并首次使兼容的高通量RNA测序成为可能。从那时起,越来越多修饰和改进的单细胞RNA测序技术被开发出来,引入了在样本收集、单细胞捕获、条形码逆转录、cDNA扩增、文库制备、测序和生物信息学分析等方面的必要修饰和改进。最重要的是,成本显著降低,而自动化和通量显著提高。
scRNA-seq的通量从几个细胞增加到数十万个细胞,而成本大大降低,这得益于scRNA-seq技术的快速发展,如基于微流控、微孔、液滴的技术以及原位条形码和空间转录组分析等(图1)。
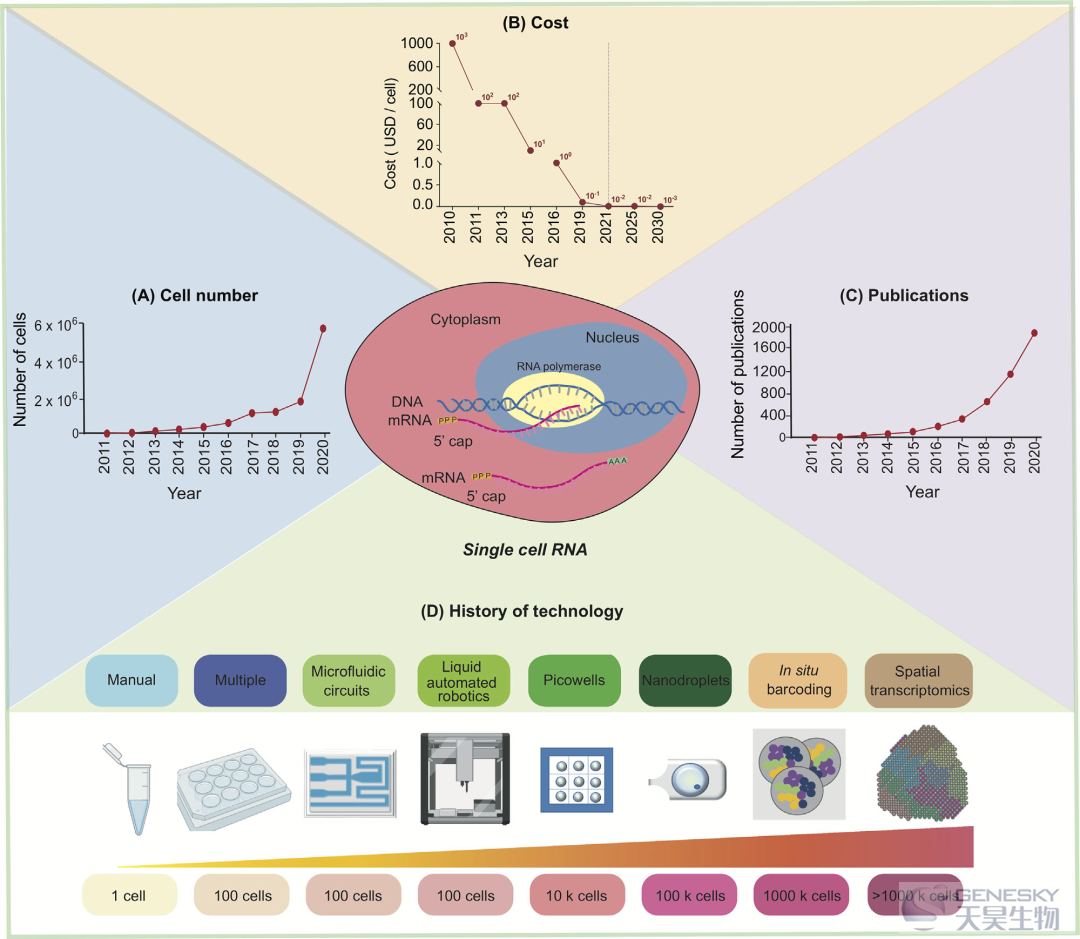
scRNA-seq的步骤主要包括单细胞分离和捕获、细胞裂解、反转录、cDNA扩增和文库制备(图2)。单细胞捕获、反转录和cDNA扩增是文库制备步骤中最具挑战性的部分。随着多种测序平台的发展,RNA-seq文库制备技术也呈现出快速而多样化的发展。因此,了解不同单细胞RNA测序文库制备方法的特点和应用,以便在科学研究中做出适当的选择,更好地将这些技术应用于临床。
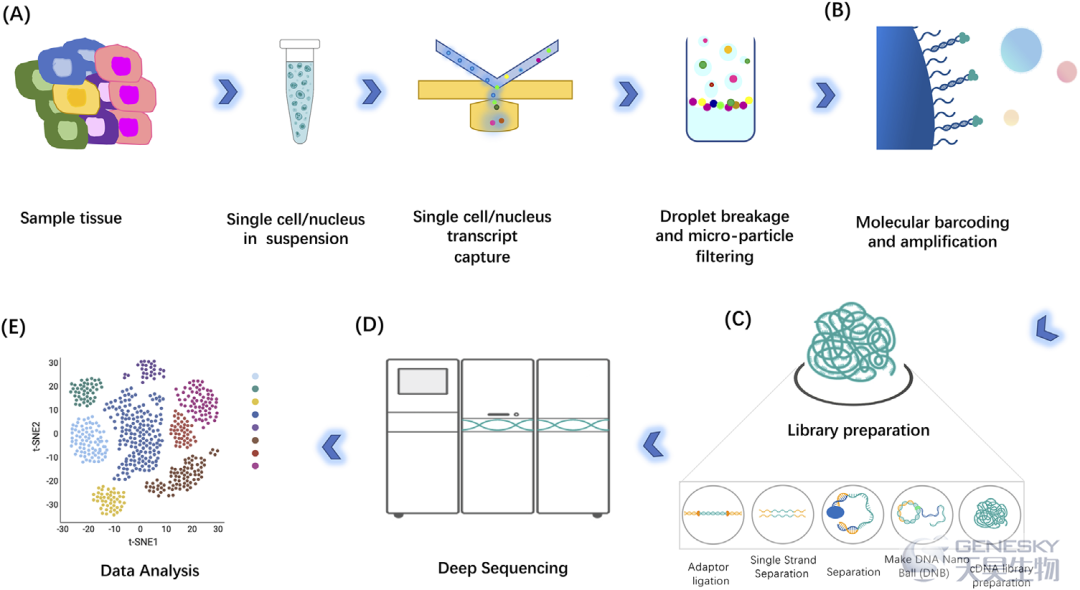
单细胞分离和捕获是从组织中捕获高质量的单个细胞的过程,从而提取精确的遗传和生化信息,并促进独特的遗传和分子机制的研究。组织块(bulk)样本传统的转录组、表观基因组或蛋白质组只能捕获来自组织/器官的整体信号,无法区分单个细胞的变异。单细胞的分离和捕获方法因生物体、组织或细胞特性而有很大的不同。细胞分离可以通过分离整个细胞、细胞特异性的细胞核或细胞特异性细胞器,甚至分离表达特异性标记蛋白的细胞来完成。最常见的单细胞分离和捕获技术包括有限稀释、荧光激活细胞分选(FACS)、磁珠激活细胞分选、微流控系统和激光显微切割。单个捕获的关键结果,特别是在高通量中,是每个单细胞被捕获在一个孤立的反应混合物中,其中单个细胞的所有转录本将被加上唯一的条形码转换为cDNA。
然而,scRNA-seq也逐渐揭示了一些固有的方法问题,如人工转录应激反应。这意味着解离过程可以诱导应激基因的表达,从而导致细胞转录模式的人为改变。这已被大量实验证实,Brink等人发现蛋白酶在37℃下的解离过程可以诱导应激基因的表达,引入技术错误,导致不准确的细胞类型识别。Adam等人也发现,37℃处的解离会导致细胞转录组的人工改变,从而导致不准确的结果。因此,建议在4℃条件下将组织解离成单细胞悬液,以最大限度地减少解离过程中引起的基因表达变化。
单核RNA测序(snRNA-seq)是一种可替代的单细胞测序方法。snRNA-seq不是对细胞质中的所有mRNA进行测序,而是只捕获细胞核中的mRNA。snRNA-seq解决了不容易分离成单细胞悬液的组织保存和细胞解离相关的问题,适用于冻存样本,与scRNA-seq相比,最大限度地减少了人工转录应激反应。snRNA-seq在许多组织类型中都非常有用,例如肌肉组织、心脏、肾脏、肺、胰腺和各种肿瘤组织,并特别适用于难以解离获得完整细胞的脑组织。Grindberg等人证实了单细胞转录组分析可以使用脑组织单个核中极低水平的mRNA。因此,snRNA-seq方法具有如下一些优势:1、与完整的细胞相比,细胞核具有容易从复杂的组织和器官中分离出来的优势(如中枢神经系统);2、snRNA-seq可以广泛应用于真核生物物种,包括来自不同界的物种;3、该方法还可以为研究核特异性的调控机制提供参考。但是需要注意的是,snRNA-seq仅捕获细胞核内的转录本,可能无法捕获与mRNA加工、RNA稳定性和代谢相关的重要生物学过程。
在将RNA转化为第一链cDNA后,得到的cDNA可以通过PCR或体外转录(IVT)进行扩增。PCR作为非线性扩增过程应用在Smart-seq、Smart-seq2、Fluidigm C1、Drop-seq、10x Genomics、MATQ-seq、Seq-Well和DNBelab C4。目前存在两种PCR扩增策略。一种采用SMART技术,利用Moloney鼠白血病病毒逆转录酶的转移酶和链转换活性,整合模板转换寡核苷酸作为接头进行下游PCR扩增。该方法是目前最常用的cDNA扩增方法。另一种策略是将cDNA的5’端与poly(A)或poly(C)连接,以在PCR反应中构建通用的接头。IVT是另一种扩增方法和线性扩增过程,用于CEL-seq、MARS-Seq和inDrop-seq方法中。它需要对扩增的RNA进行额外一轮的逆转录,这会导致额外的3’端覆盖偏倚性。这两种方法都可能导致扩增偏倚。为了克服扩增相关的偏倚性,在反转录步骤中引入独特的分子标记(UMI)对细胞内的每个单个mRNA分子加上条形码,从而提高scRNA-seq的定量性质,并通过有效消除PCR扩增偏倚提高了读取精度。
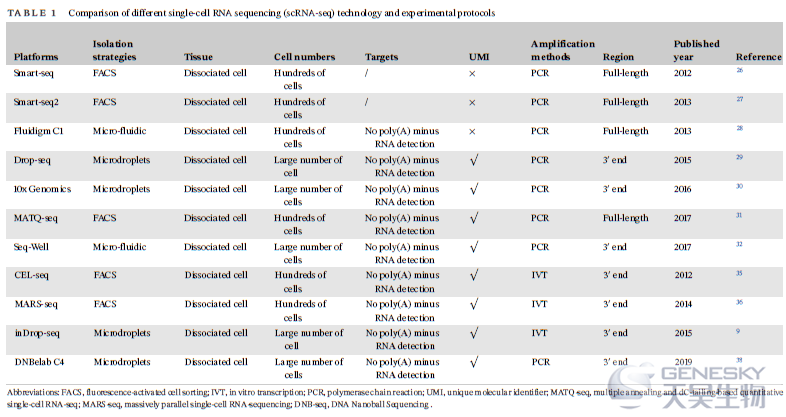
总之,我们想强调一下在准备scRNA-seq文库时应该强调的问题:(1)如何从总RNA中捕获感兴趣的RNA类型,也称为RNA富集;(2)如何将RNA反转录成合适大小的cDNA;(3)如何将接头连接到cDNA末端。此外,在scRNA-seq文库制备的过程中,还有一些剩余的挑战需要克服。例如,细胞间的高可变性通常发生在scRNA-seq数据中,这是由细胞中RNA捕获和随机转录的技术差异引起的。此外,由于高测序成本,以前的scRNA-seq方法只集中在转录组的5'或3'末端。事实上,单细胞RNA-seq的区域样本应该取决于实验目的。例如,尽管3’端测序比全长测序便宜,并且可以通过添加非模板polyA尾来提供3’的最佳编码区数据,但它不能对整个尾序列进行测序,也不能明确报告尾部所附着的mRNA异构体。此外,scRNA-seq文库制备的质量还受到其他因素的影响,如技术噪音、生物噪音等。技术因素包括RNA捕获效率和质量、文库制备过程中的随机缺失、单细胞扩增技术和实验批次效应。生物噪音方面,生物标本的性质和不同的遗传背景(如细胞大小、基因表达)以及环境的动态随机变化(各种细胞状态、细胞周期状态)难以通过实验操作加以控制。因此,在准备scRNA-seq文库的过程中,如何最大限度地减少RNA损失和最大化信息精度仍然是一个关键的挑战。
对scRNA-seq数据的分析是另一个关键因素,也是现在的主要需求,以扩大该技术在生命和临床科学中的应用。为了确保单细胞转录组分析工具的可用性,许多开发人员已经做出了相当大的努力。到2021年5月28日,已经开发并提供了近1000种不同的生物信息学工具。单细胞转录组分析工具数量的增加说明了分析方法在该领域的重要性,但这也意味着选择单细胞数据分析工具的更多困惑。在本节中,我们根据关键步骤回顾了单细胞转录组分析的基本过程(图3)和分析模块,其中也涵盖了基因水平和细胞水平的探索性分析。对于高级的单细胞转录组数据分析,可以参考原文附表中总结的特定工具及原始文章。
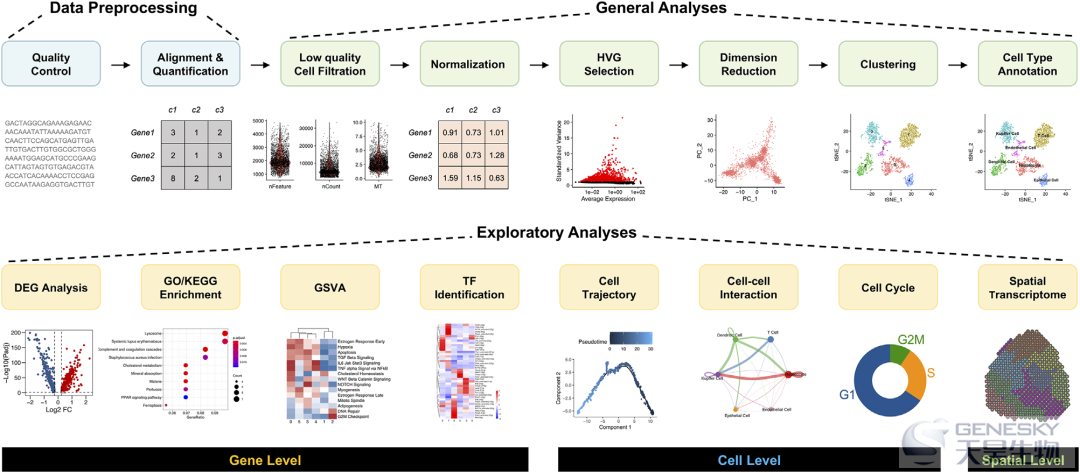
3.1 数据预处理
单细胞转录组原始测序数据的基本格式包括FASTQ和BCL格式,这些格式依赖于数据源和测序平台。由于只有FASTQ文件可以直接实现质控,一旦原始数据不是FASTQ格式,第一步就是用适当的工具将其转换为FASTQ格式。可以使用cellranger mkfastq从BCL文件生成FASTQ文件。重要的是,除了BCL文件的路径之外,还应该提供一个简单的CSV矩阵文件,至少包括三列(lane、sample和index)。然后,FastQC可以用于评估原始单细胞RNA测序数据的质量。
高质量的片段(reads)需要使用适当的比对器(如STAR或Tophat)映射到特定的参考基因组。计数是Cell Ranger最重要的功能,其内部包含了比对、过滤、UMI计数和其他实际步骤。Cell Ranger使用了一个称为STAR的比对器,该比对器对基因组的片段进行剪接感知比对,然后使用转录注释一般转移格式(GTF)文件根据片段是否准确地与基因组比对,将这些片段分类为外显子、内含子和基因间区。
3.2 一般性分析
在单细胞悬液的制备过程中,由于不可避免的自然现象、实验操作和技术障碍,活细胞可能会死亡、细胞膜损伤或多细胞粘附。为了消除低质量细胞对基因表达的干扰,需要使用合适的工具进行第二轮质控,如Seurat、scran和scanpy。在引用方面,Seurat是最受欢迎的,它内置了处理低质量细胞过滤的功能。一个细胞是否需要保留,基本上可以通过以下质控指标来判断:基因的数量,UMI(转录本)的数量,线粒体基因的百分比,核糖体蛋白基因在每个细胞中的百分比。过滤阈值的设置没有绝对的标准,它通常取决于被分析的细胞和组织的类型。比如有研究过滤掉≤100或≥6000个表达基因、≤200个UMIs和≥10%线粒体基因的细胞;也有设置保留200<表达基因<2500、300
与分析传统的bulk RNA-seq数据类似,在分析单细胞RNA测序数据时,每个细胞都被视为一个独立的样本。原始表达矩阵不能直接用于下游分析,因为由于系统错误或技术噪音(如每个细胞的测序深度和转录组捕获率的差异),细胞之间的表达水平无法进行比较。标准化旨在抵消技术噪声或偏差,并确保每个细胞之间的可比性。在2020年,Lytal等人评估了七种归一化方法的有效性,包括BASiCS、GRM、Linnorm、SAMstrt、SCnorm、scran和Simple norm。值得注意的是,Linnorm和scran的速度优势来自于用C++编写,并在R中实现,适合于大数据集。相反,BASiCS和SCnorm需要更长的时间来生成更精确的结果。总的来说,这些方法之间存在很大的差异,不同的工具在不同的情况下表现最佳。
单细胞RNA测序数据集是高维的,一个样本中有数万个细胞,每个细胞中有数千个基因表达。每个细胞中的大部分基因都属于看家基因,因为它们的特点是细胞之间的表达水平没有显著变化,它们的存在往往会掩盖真正的生物信号。在数据集中表现出高度细胞间变异的特征子集也被称为高度可变基因(HVGs)。HVGs不仅突出了生物信号,而且由于计算量的显著减少,大大加快了对单细胞RNA测序数据下游分析的速度。一个高质量的HVGs应该包含能够区分不同细胞类型的基因,HVGs的质量对聚类的精度有显著影响。2018年,Yip等人评估了七种检测HVGs的方法,包括BASiCS、Brennecke、scLVM、scran、scVEGs和Seurat,发现不同方法的聚类结果以及运行时间存在很大差异。与其他方法相比,scran可以检测出稳定数量的HVGs,并且具有良好的运行时间。Brennecke在大范围数据集上具有稳定一致的性能。scran和Seurat在处理部分数据集时表现最佳。BASiCS和sclvm_logvar比其他的要慢得多。
不同的scRNA-seq数据可能产生于不同的时间、不同的测序平台,这些数据之间不可避免地存在着技术上或非生物学上的显著批次效应。scRNA-seq数据中的批次效应一直困扰着下游分析,因为它可以破坏基因表达模式,然后导致错误的结论。因此,批次效应校正对于分析scRNA-seq数据至关重要。虽然已经提出了一些针对scRNA-seq数据的批量效应校正算法,如Scanorama和Seurat V4,其只能一次合并两个数据集,并通过迭代整合多个数据集。它们中的大多数会消耗大量的计算内存和时间,并且随着scRNA-seq数据数量的增加,这一需求很可能会增加。最近,Zou等人提出了一种新的基于深度学习的方法,称为deepMNN,以纠正scRNA-seq数据中的批次效应。它比较了deepMNN和最先进的批次校正方法的性能,包括广泛使用的Harmony、Scanorama和Seurat V4方法,以及最近开发的基于深度学习的MMD-ResNet和scGen方法。结果表明,deepMNN的精度优于现有的常用方法,特别是在大数据集情况下。而deepMNN算法的时间复杂度和空间复杂度几乎是优良的。对大数据集完成批次效应校正耗时17 min,而Harmony和Scanorama分别耗时35和77 min。此外,它具有比Seurat V4和scGen更大的存储空间。同时,deepMNN可以在一步中整合多批数据集,无需多次迭代。deepMNN的这些特性使其有可能成为大规模单细胞基因表达数据分析的新选择。
除特征选择外,降维也是处理这类高维数据的主要策略之一。对于单细胞RNA测序数据,通常需要进行两轮降维,首先进行主成分分析(PCA)降维,然后进行t-分布式随机近邻嵌入(t-SNE)或统一流形近似与投影(UMAP)降维可视化。PCA是一种数学线性维度算法,它利用正交变换将一系列可能线性相关的变量转换为新的线性不相关的变量,从而利用新的变量在低维上显示数据的特征。PCA已广泛应用于scRNA-seq研究,以克服任何单一特征中广泛的技术噪音。Wu等在2019年对这两种非线性降维方法进行了系统的比较。他们指出UMAP在高维细胞学和单细胞RNA测序中的使用,特别强调与t-SNE相比UMAP表现出运行时间更快和一致性,以及更有意义的细胞群结构和连续体的维持。此外,UMAP在细胞子集的连续性方面比t-SNE有明显的优势,因为它保留了更多的全局结构,尽管t-SNE仍然应用于许多单细胞研究,似乎是由于更好的视觉偏好。
单细胞RNA测序数据的复杂性促进了广泛的聚类方法的发展。基于恢复已知亚群的能力、稳定性、运行时间和可伸缩性,最近的一篇论文在总共12个不同的数据集上评估了14种聚类方法。值得注意的是,SC3和Seurat在这些方法中综合来看表现更好,Seurat的速度要快几个数量级。在cluster数量相同的情况下,Seurat通常与真实区分的一致性最好,而FlowSOM在cluster数量大于真实数量时其与真实区分的一致性更好。
聚类后,为每个聚类分配生物学注释是后续分析的基础。通常,在scRNA-seq数据中注释细胞的工作流程包括三个主要步骤:自动注释、人工注释和湿实验验证。首先,主要的自动化注释工具利用一组预定义的标记基因,这些标记基因在已知的细胞类型中特异表达,通过将它们的基因表达模式与已知的细胞类型匹配来标记cluster。自动细胞标注方法的优点是快速、可重复性好,对常见细胞类型的标注结果更可靠。然而,由于参考标记基因集的限制,它无法定义罕见的和新的细胞类型。2020年,Huang等人对Seurat、scmap、SingleR、CHETAH、SingleCellNet、scID、Garnett、SCINA、CP和RPC等10种细胞类型注释方法进行了系统的比较和评估。他们发现,在Seurat、SingleR、CP、RPC和SingleCellNet这五种最常用的方法中,Seurat是注释主要细胞类型的最佳方法。然而,Seurat在预测罕见细胞类型和区分高度相似的细胞类型方面表现相对较差。其次,人工注释是标注细胞的金标准,虽然它需要搜索相关文献和挖掘已有的scRNA-seq数据,既主观又费力。最后,通常需要湿实验来进一步验证scRNA-seq的发现。传统的验证方法包括免疫荧光和免疫组化,这两种方法都是基于抗体与抗原(标记基因编码的表面蛋白)特异性结合的原理来证明数据分析得到的细胞类型的真实存在。此外,新兴的空间转录组测序技术也可以考虑提高注释的可靠性,它可以结合细胞成像和scRNA-seq在一个实验中检测空间转录模式和细胞形态。
为了准确地揭示特定细胞群体的功能偏倚和生物学意义,有必要对目标差异表达基因集进行功能富集分析。功能富集的通用分析策略也适用于单细胞数据,如GO和KEGG通路。大量成熟的功能富集分析工具已经开发出来。Huang等人在2009年权衡了68种富集分析工具的优缺点后,综合比较了它们。此外,GSVA还以通路为中心的方式广泛应用于功能富集分析等标准分析中。GSVA可以计算每个样本中不同信号通路的富集分数,以评估表型差异的原因,可以作为KEGG通路的补充,使结果更具生物学解释性。
为了从scRNA-seq数据中识别每个细胞簇中富集的转录因子,Aibar等人于2017年开发了SCENIC可以实现转录因子(TF)的推断,首先通过搜索靶基因的假定的调控区域来富集转录因子基序。然后转录因子基序富集可以实现候选TF调控因子与候选靶基因的连接。虽然SCENIC可以用R和Python实现,但是强烈推荐使用pySCENIC来运行大数据集,因为它可以更快地实现SCENIC流程。值得注意的是SCENIC的最新版本支持智人、家鼠和黑腹果蝇,并有可能手动创建其他物种的定制数据库。虽然由于其出色的可扩展性和对各种数据库的稳健性而被广泛使用,但它忽略了不同细胞类型中基因调控机制的动态变化。2020年,Ma等人从scRNA-seq中开发了IRIS3,一个整合的细胞类型特异性调控推断服务器。在实际应用中,IRIS3更适合没有大量编程技能的研究人员使用其用户友好的web服务器。然而,IRIS3需要在准确性和效率上不断改进。
拟时分析可以在单细胞水平上推断细胞的轨迹,有望发现罕见的细胞类型和隐蔽的状态。在拟时分析方面,已经开发了不同类型的分析工具。2019年,Saelens等人对45种拟时分析工具进行了综合比较,发现现有工具具有很强的互补性。Monocle是应用最广泛的拟时分析工具之一,它借鉴显式主图来描述数据,并通过嵌入反向图来重建单细胞轨迹,以提高预测轨迹的稳健性和准确性。着重指出,建立单细胞基因表达动力学的整个过程在很大程度上是数据驱动的。
生物体受到刺激后会自我调节以维持体内稳态,这需要多种细胞的共同参与和协调。随着细胞-细胞通讯研究的快速发展,分析细胞-细胞通讯的工具不再有限,包括CellChat、CellPhoneDB、NicheNet、SingleCellSignalR和iTalk等。虽然每一种工具都依赖于细胞表面配体和受体相互作用的强度,但每一种工具都有其优缺点。具体来说,如果要考虑配体和受体的结构组成,CellPhoneDB是首选。如果需要考虑辅助因子(如启动子和拮抗剂)的调控,可以选择CellChat来提高性能。还建议灵活地结合多种细胞-细胞通信分析工具,以避免系统偏差。
单细胞悬液中的每个细胞都处于细胞周期的特定阶段:DNA合成前期(G1期)、DNA合成期(S期)、DNA合成后期(G2期)或有丝分裂期(M期)。每群细胞都混合着不同细胞周期的细胞。Seurat的CellCycleScoring功能根据其内置包内的G2/M和S期标记基因的表达给每个细胞打分。近年来,基于机器学习的方法已经被开发出来,从单细胞RNA测序数据预测细胞周期阶段。2015年,Scialdone等人比较了五种已建立的监督机器学习方法,以及基于转录组将细胞分配到其细胞周期阶段的定制预测器。他们特别指出,只有基于PCA的方法和定制的预测器性能最好,可以稳健地捕获细胞周期信号。
虽然我们解释了单细胞测序分析过程的主要步骤,但还有很多其他重要的方面值得更多的关注和探索,如scRNA-seq和CRISPR筛选的联合应用,scRNA-seq和多组学的综合分析,包括scATAC-sEquation(单细胞染色质可及性和转录组测序)、scMT-sEquation(单细胞甲基化组和转录组测序)、CITE-sEquation(通过测序对转录组和表位进行细胞索引)和空间转录组。这些技术的结合可以更好、更深入地了解关键的生物过程和机制,是未来单细胞技术发展的一个重要方向。在单细胞RNA转录组研究领域,分析算法和工具在改善数据探索和更好地理解细胞功能方面仍有很大的潜力。因此,我们也鼓励读者阅读其他关于scRNA-seq分析各个方面的优秀综述,以获得更多的灵感。
迄今为止,单细胞RNA表达谱正迅速成为包括人类、动物和植物在内的各种研究的不可替代的方法,使之能够前所未有地更准确、快速地识别组织中罕见和新颖的细胞(图5)。此外,拥有mRNA和蛋白质水平上基因表达、代谢物、细胞间通讯和空间图谱的信息,使得解决健康和疾病中细胞组成和功能的谜题成为可能。虽然scRNA-seq的最初发现和使用主要是在动物和后来的人类细胞上进行的,但植物科学中的测序仍处于早期阶段,还有许多令人兴奋的挑战有待克服。
到目前为止,由于技术上的挑战或关于细胞类型和发育生物学发现的信息非常有限,scRNA-seq的应用仍然仅限于少数植物。一些植物研究课题组使用了分子遗传学中最常用的模型植物拟南芥根进行了高通量的scRNA-seq和空间转录组学分析,由于其具有相对较少的细胞数量、已知的基因标记和通过酶消化细胞壁降解分离单个细胞的简单方法。在拟南芥根的成功应用后,研究越来越多地应用于拟南芥的其他部分和其他植物物种的研究,如水稻的叶和根,番茄和玉米。此外,随着人类细胞图谱的建立,植物科学界于2019年发起了植物细胞图谱联盟,旨在收集有关各种植物细胞类型及其核酸、蛋白质和代谢物的更多信息。关于植物scRNA-seq数据的各种基于网站的图片信息可以在下边网站上找到:https://www.zmbp-resources.uni-tuebingen.de/timmermans/plant-single-cell-browser。然而,快速发展的植物单细胞生物学领域还可以提供更多,包括整合测序scRNA-seq、snRNA-seq和空间转录组学、成像技术和组学将有助于进一步了解单细胞水平上基因型的变化。
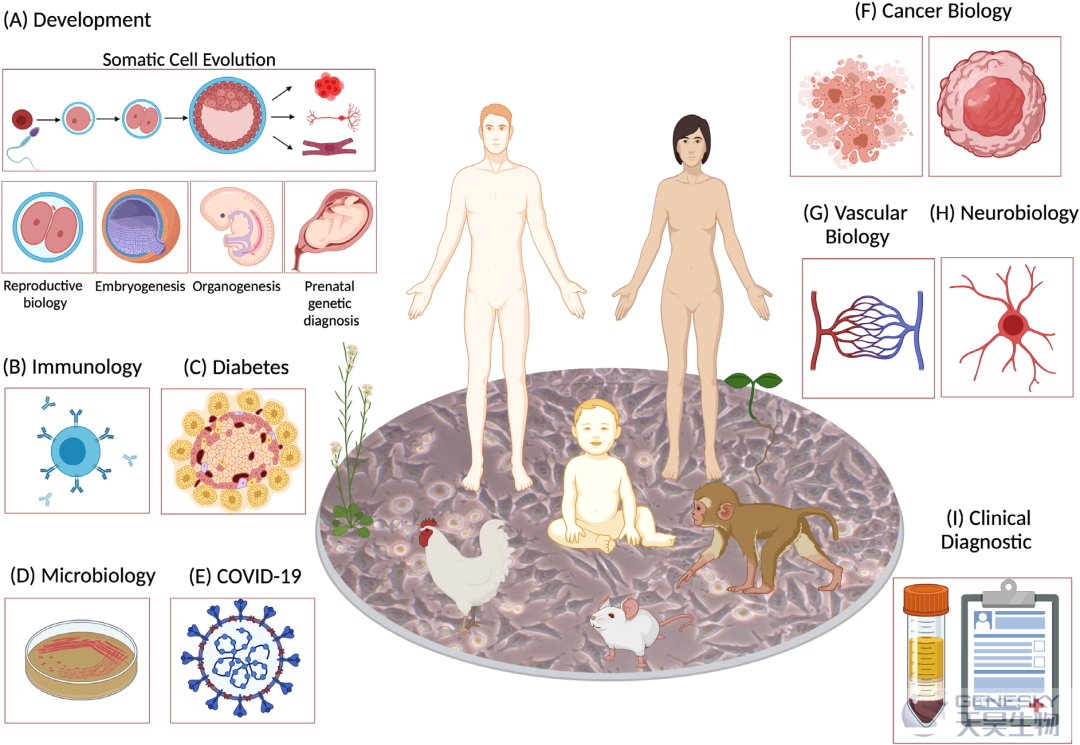
scRNA-seq成为一种强大的工具,用于分析、识别、分类和发现来自不同人体器官和组织的新的或罕见的细胞类型和亚型,提供发育、免疫学、糖尿病、微生物学、Covid- 19、癌症生物学、血管生物学、神经生物学、临床诊断和许多其他学科健康和疾病状态下更深刻的信息(图5)。
每个组织/器官都包含许多形态和功能上不同的细胞群,处于不同的状态,生理转变,分化轨迹和空间位置。这种复杂但同步良好的微环境保持着内稳态,直到出现可能将正常细胞结构转变为肿瘤的极端情况。为了了解肿瘤的起源、细胞的进化起源、肿瘤的进展、转移和治疗反应,进一步认识具有必需的免疫和基质浸润的肿瘤微环境是十分重要的。scRNA-seq分析可以区分功能健康的细胞和肿瘤不同发育阶段的癌细胞。这使得通过识别和确定对不同药物的敏感性来进行更精确的预后和诊断,并为癌症制定最有效的治疗策略。最初,scRNA-seq技术专注于分析器官的单个部分,其异质性和涉及的细胞类型,从而产生全面的数据。虽然有许多关于肿瘤的单个部分的scRNA-seq报告,其异质性和涉及的细胞类型,但每种细胞类型的生物学功能是什么,以及细胞如何相互交流和协同来完成它们的任务仍然是很大的挑战。大多数的困难来自于肿瘤组织在体内不同的位置。因此,它们的微环境包含了处于不同状态和阶段的各种肿瘤细胞和非肿瘤细胞。此外,如果在不同的时间和条件下获取组织,即使在同一个肿瘤切片内,细胞样本的混合和比例也可能会有很大的不同。此外,单细胞基因表达数据往往包含大量噪音,因此,同一类型的细胞可能最终在不同的簇中,而不同类型的细胞可能由于批次效应而在同一簇中。因此,在计算细胞类型特异性参考矩阵之前,需要仔细挑选出高质量的细胞簇。虽然scRNA-seq非常有用,但RNA表达检测并不总是提供蛋白质水平或翻译后修饰的信息。最近,scRNA-seq研究得到了其他技术的支持,包括质谱流式(飞行时间流式细胞术,CyTOF),例如,两项研究都证实了肿瘤中的调节性T细胞(T-reg)比血液或邻近正常组织表达更高水平的肿瘤坏死因子受体超家族成员9(TNFRSF9),诱导T细胞共刺激因子(ICOS)和细胞毒性T淋巴细胞相关抗原4(CTLA4),可能反映了一种激活状态。此外,通过将空间信息添加到scRNA-seq数据中,我们能够理解分子、细胞和空间组织的结构以及原位细胞间的相互作用。
肿瘤微环境被免疫细胞类型浸润,即T淋巴细胞、CD8+ T细胞、肿瘤相关的巨噬细胞、肿瘤相关的成纤维细胞、上皮细胞和肿瘤干细胞,但免疫反应的类型及其对肿瘤生长的影响,转移和死亡在不同的癌症和不同的肿瘤之间差异很大。免疫细胞既具有抑制和杀死肿瘤细胞的抗肿瘤作用,又具有促进肿瘤生长和免疫逃逸的促肿瘤活性。迄今为止,免疫细胞的改变在肺癌、乳腺癌、头颈部鳞癌、鼻咽癌、头颈部癌、胰腺癌等肿瘤中都有较先进的方法报道。
在肿瘤中,不同类型的细胞,包括肿瘤细胞,通过各种信号通路进行活跃的通信。识别肿瘤和非肿瘤细胞之间的通信将为新的治疗策略的发展提供重要的见解。也有一些计算方法被开发出来,从scRNA-seq数据推断细胞间的通讯,如SingleCellSignalR, iTalk和NicheNet,通常只使用一个配体和一个受体基因对,CellPhoneDB v2.0通过考虑异聚复合物成员的最低平均表达来预测两个细胞群之间丰富的信号相互作用。另一个平台CellChat预测细胞的主要信号输入和输出,并使用网络分析和模式识别方法对功能进行信号协调。
可通过scRNA-seq研究的肿瘤异质性的另一个重要方面是肿瘤形成的进化过程,该过程已被发现在肿瘤形成以及诸如化疗和耐药性等特征的获得中发挥重要作用。异质性的持续积累可能反映了癌症的演变,而scRNA-seq可以为复杂肿瘤内的少数抗治疗细胞群提供有意义的见解,可以用于根据肿瘤类型选择合适的治疗方法,并更精确地治疗个体患者。黑色素瘤、肝癌、胶质母细胞瘤、乳腺癌、前列腺癌的研究将多种信息整合在单个癌细胞中,破译了癌症异质性和进化的秘密。此外,另一项新兴技术,如空间转录组测序,整合了细胞的空间位置信息,提供了基因表达异质性的信息。类器官可以模拟一些肿瘤的异质性,3D组织可以用于药物筛选。所有这些技术对于正确理解肿瘤生态系统的3D体积,内皮细胞在肿瘤中的作用,肿瘤中血管生成的发展以及肿瘤对不同治疗的反应都是必要的。
scRNA-seq技术的另一个重要应用是更好地了解糖尿病中β细胞的发育和病理。1型糖尿病(T1D)的治愈在于β细胞的恢复。然而,要产生有功能的β细胞,需要对胰腺发育、其分子事件及健康和疾病中的细胞异质性有广泛的了解。scRNA-seq对发育中的胰腺的研究首先在小鼠模型上进行,随后最近对人类多能模型的研究主要是在胚胎干细胞(ESCs)和诱导多能干细胞(iPSCs)3D模型上进行。在小鼠模型上进行的研究揭示了胰腺发育的几个重要方面。一些研究小组开始揭示发育阶段的基础知识,在野生型模型上鉴定了一种新的α-细胞特异性标记Slc38A,另一组和Zeng等人发现了不同的β细胞异质性、出生后β细胞成熟的转录动态和出生后β细胞增殖。
此外,与单层(2D)细胞培养相比,3D细胞培养微环境更类似于体内胚胎发生和器官发生。这种新方法还增加了hiPSC来源β细胞的功能。值得注意的是,3D类器官被用作患者特异性细胞模型,为研究T1D转录组提供了替代平台。CRISPR-Cas9基因编辑技术增加了基因工程hiPSCs的可及性,允许操纵已知或假定的发育调控因子,以评估其在人类组织中的功能。此外,scRNA-Seq可以与CRISPR或谱系示踪结合使用。尽管有这些巨大的希望,重要的是要注意,由于外分泌细胞的高水解酶含量,研究单个胰腺细胞特别具有挑战性。克服这些局限性的方案正在不断发展,包括解剖胰腺的快速冷冻,然后是snRNA-seq。此外,scRNA-seq以外的单细胞组学技术已经开发出来,如Patch-Seq。
除了scRNA-seq在基础生命科学研究中的应用,这项技术也被证明是了解传染病的有力工具。截至2021年11月3日,由冠状病毒SARS-CoV-2引起的2019冠状病毒病大流行已影响到全球2.48亿多人。了解COVID-19感染的发病机制,对于预防传播、降低感染严重程度、快速有效地制定新的治疗策略具有重要意义。到目前为止,已经开展了大量使用单细胞RNA测序技术的研究,以了解COVID-19患者的免疫细胞分布和反应,并发现临床结果因年龄、性别、严重程度和COVID-19疾病阶段的不同而不同。他们发现,与健康对照组相比,COVID-19在人类中诱导了一种独特的免疫细胞信号,特别是在早期恢复阶段。通过对COVID-19患者及健康人群外周血进行scRNA-seq检测,发现中度、重度、恢复期患者与对照组关键免疫细胞组成的差异。COVID-19患者的大多数细胞类型表现出强烈的干扰素α反应和急性免疫反应。除了外周血单核细胞(PBMC)和支气管肺泡灌洗液(BAL)免疫细胞的单细胞测序,COVID- 19组织/器官的单核RNA测序也为疾病的严重程度和进展提供了重要的病理学见解。通过对COVID-19患者24例肺、16例肾脏、16例肝脏和19例心脏解剖组织样本的单细胞测序及14例肺样本空间转录组测序,Delorey等人揭示了严重SARS-COV-2感染的生物效应及患者肺上皮、免疫和基质的重塑。大流行还远未完全消失,而scRNA-seq无疑仍将是一个重要的工具,准确揭示全球不同变体的免疫反应。综上所述,单细胞RNA测序技术在抗击COVID-19的斗争中获得了更科学的见解,未来不仅可以用于当前的SARS-CoV-2,还可以与传统方法结合用于其他病原体的检测。
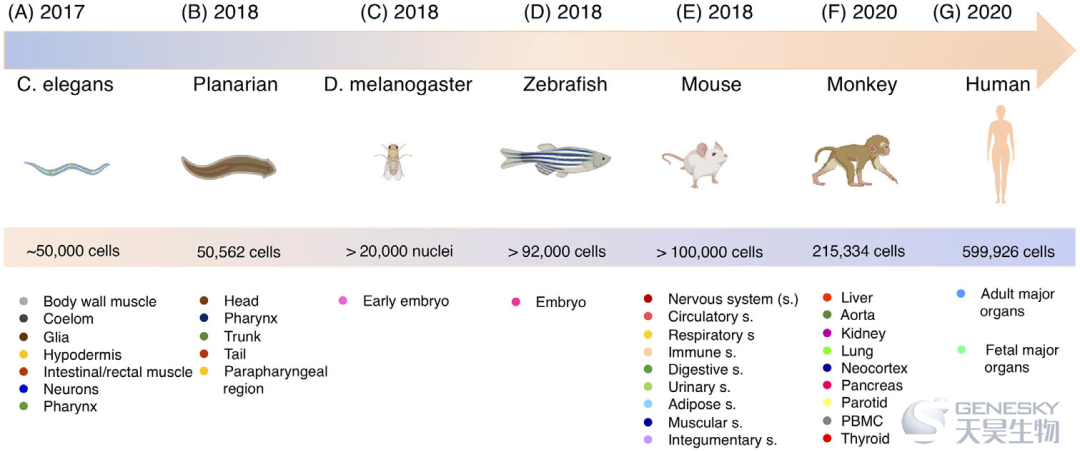
单细胞RNA测序技术在过去十年中已被证明是生命科学领域的一项革命性技术。高通量单细胞RNA测序技术和计算工具的发展,使得该技术在生命科学中的几乎所有应用领域都可获得和应用。单细胞RNA测序技术的一个重要基础知识是在组织、器官和有机体中构建单细胞图谱。为此,在研究和建立细胞图谱方面作出了相当大的努力。仅提几个,全身单细胞图谱已经应用于秀丽隐杆线虫、涡虫、黑腹果蝇、斑马鱼、小鼠、猕猴和人类(图6)。随着技术和应用的发展,预计将产生越来越多的单细胞RNA测序数据,并将其集成到一个可公开访问的数据库中,以促进了解基因和细胞在健康和疾病中的功能。
结合scRNA-seq和其他大型遗传筛选工具将进一步扩大该技术的应用。其中一种组合技术是将scRNA-seq和基于CRISPR的基因组范围遗传筛选相结合,例如Perturb-seq,它可以评估用CRISPR敲除几个基因的转录效应,而LinTIMaT集成单细胞转录组数据和突变数据进行谱系追踪。除了CRISPR介导的突变,也有可能将scRNA-seq与CRISPR介导的基因激活或干扰相结合。这些组合应用使我们能够大规模地研究基因对细胞转录组和功能的影响。随着单细胞RNA测序和CRISPR基因编辑技术的不断发展,预计还会有更多这样的组合技术和应用出现,有助于更好地理解基因和细胞功能。
本文就单细胞RNA测序技术及其应用进行综述。然而,应该指出的是,单细胞测序技术已经发展到可以检测几乎所有的组学,如单细胞全基因组测序、单细胞拷贝数变异测序、单细胞表观遗传标记(即DNA甲基化、染色质可及性)测序,单细胞蛋白组和单细胞代谢组。越来越多的多组学研究和分析有望全面描述健康或疾病状态下细胞类型的基因调控过程、功能、分子和相互作用。
尽管有这些巨大的希望,单细胞RNA测序的一个主要缺点是组织学信息的丢失,因为单细胞和单核悬液都必须从组织中制备。虽然轨迹分析可以帮助预测不同细胞类型之间的关联和过渡,但与组织消化、细胞解离和保存相关的其他混杂因素可能会改变基因表达和细胞表征。单细胞转录组学的空间维度是在分子水平上研究整个生物体结构的一个重要步骤和突破。一些空间转录组学方法已经被开发出来,并在概念验证研究中得到证明,例如基于条形码阵列的显微切割组织转录本捕获和原位测序。2020年,空间分辨转录组学技术被Nature Methods选为年度方法。从空间和时间上揭示复杂组织和器官中的单细胞转录将成为了解组织/器官/有机体中细胞的组成、复杂性、相互作用和功能的正在崛起的转化工具。
未来另一个有前景的应用是将scRNA-seq技术整合到常规临床诊断和个性化医疗中。然而,目前大多数基于scRNA-seq的临床研究仍处于探索阶段,主要侧重于重新审视和更好地理解疾病过程,识别诊断和治疗标志物。尽管每个细胞的成本已经显著降低,但每个样本的成本(包括文库制备和测序)仍然很高(图1)。这仍然是使用scRNA-seq作为常规诊断工具的一个限制因素。其他剩下的挑战是scRNA-seq数据处理、分析、展示和解释。为了进一步拓宽基于scRNA-seq的临床应用,需要具有用户友好界面的自动scRNA-seq数据分析流程,最重要的是,它可以供没有任何生物信息学技能和背景的人员使用。一个这样的例子是来自Galaxy Community(https://galaxyproject.org/use/singlecell/)的单细胞组学工作台,它集成了20多个生物信息学工具。由于大量的开源工具已经被开发出来,更精简和自动化的scRNA-seq数据分析和可视化平台有望在未来产生并可用。总之,这篇综述简要介绍了单细胞RNA测序技术及其应用。该技术的不断发展将扩大其在临床和个性化医疗中的应用。
咨询沟通请联系
18964693703(微信同号)

创新基因科技,成就科学梦想
 咨询热线:400-065-6886
咨询热线:400-065-6886

